
Research Highlights
How cells control their protein content: An introduction

Cellular protein levels are determined by both synthesis and degradation. In eukaryotic cells, the Ubiquitin-Proteasome System controls most selective protein breakdown, involved in most aspects of cellular activities. Mis-regulation of protein degradation is widely associated with human pathology, such as cancer, dementia, muscle dystrophy, autoimmunity, aging and etc.
Most proteins can be ubiquitylated, which is catalyzed by ~600 ubiquitylation pathways covalently attaching ubiquitin, a 76-amino-acid small protein, to lysine residues on the targets and ubiquitin itself. The resulting complex patterns of ubiquitin form an enigmatic “ubiquitin code” and dictate the consequence of this modification. Various effector proteins, including the proteasome, recognize ubiquitylation patterns and execute its instructions to regulate the selectivity, rate and manner of protein degradation.
Our work focuses on three main questions:
How does the ubiquitylation patten control the specificity and rate of protein degradation?
How are protein aggregates, as well as other difficult substrates, recognized and cleared by the degradation system?
How can we rewire the degradation system to treat diseases?
I. A Size Filter in Protein Quality Control
Size-dependent phenomena are universal in biological systems, though few biological mechanisms account for size selectivity. During protein degradation, the proteasome processes most soluble substrates but typically cannot degrades protein aggregates which are instead sequestered into a perinuclear deposit, called “the aggresome”, via dynein transport. Aggresome formation critically complements the canonical degradation pathways when their activities are compromised. How does the transport machinery distinguish aggregates vs. soluble substrates and only sequester the former in aggresome?
Most previous studies on aggresome have relied on cell models. These systems face strong limitations due to the complexity of the intracellular environment, which can be exacerbated under proteotoxic conditions. In this study, we developed a novel in vitro reconstitution system for studying aggresome formation using Xenopus laevis egg extract and a chemically-inducible aggregation-prone protein AgDD. Our reconstitution system allowed us to study in detail the interactions between the dynein motor and misfolded protein aggregates of different sizes. Unlike conventional dynein-mediated transport, which favors small cargoes due to the higher viscosity drag experienced by larger cargoes, we found that dynein-mediated transport of protein aggregates requires the aggregate to have a diameter of at least ~0.4µm. Above this threshold, transport is slow but the velocity positively correlates with aggregate size, unlike conventional cargoes, which typically show a negative size selectivity. We were able to provide a rationale for this observation by showing that the aggresome-dynein adaptors have unusually weak interactions with dynein, leading to recurrent disassembly and reformation of the cargo-dynein-microtubule complex. Through mathematical modeling, we showed that this disassembly-reformation process biases larger aggregates in the active transport state, quantitatively accounting for the aggregate preference by the aggresome pathway. This study is a salient example of how basic physical principles are utilized by biological systems to accomplish important functions. The positive size selectivity not only establishes the basis for the substrate selectivity in aggresome formation but also has important implications in the mechanism of neurodegeneration.
Related publication:
Rui Fang, Luolan Bai, Mengying Zhou, Kevin Dong, Boyan Li, Tim Mitchison, Ying Lu (2025). Episodic transport of protein aggregates achieves a positive size selectivity in aggresome formation. Nature Communications (in press). Preprint available from: https://doi.org/10.1101/2024.08.06.606767
II. “Magic Eraser” in the Degradation System
Related publication:
Mengying Zhou, Rui Fang, Louis Colson, Katherine A. Donovan, Moritz Hunkeler, Yuyu Song, Can Zhang, Siyi Chen, Dong-hoon Lee, Gary A. Bradshaw, Robyn Eisert, Yihong Ye, Marian Kalocsay, Alfred Goldberg, Eric S. Fischer, Ying Lu (2025). HUWE1 Amplifies Ubiquitin Modifications to Broadly Stimulate Clearance of Proteins and Aggregates. BioRxiv 542886 [Preprint]. Available from: https://doi.org/10.1101/2023.05.30.542866 (in revision)
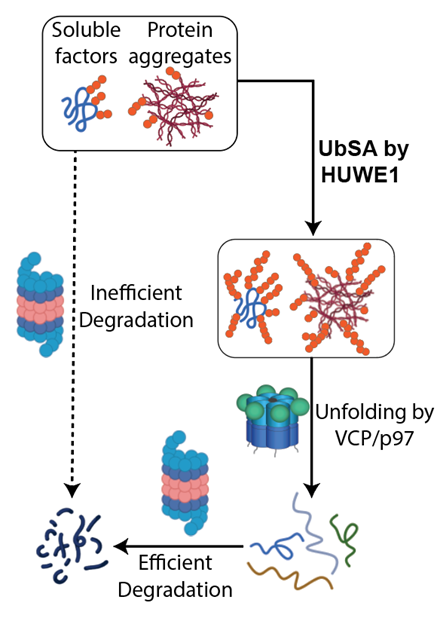
Although the proteasome is involved in degrading most cellular proteins, we found during studying the energy landscape of proteasome that the proteasomal ATPase complex exerts only ~1.2kcal/mol mechanical energy per power stroke, too low to unfold a typical well-folded domain such as GFP for proteolysis in the core particle. This finding primed the “Difficult Substrate Hypothesis”, suggesting that the degradation system recruits additional factors or alters its reaction sequence in order to degrade certain substrates, namely structurally-stable proteins, protein aggregates and some membrane proteins, that are otherwise hard to degrade by the conventional degradation machinery.
Following this hypothesis, we looked for factors empowering the degradation machinery. In a recent study, we discovered a HECT-family ubiquitin ligase HUWE1 is broadly involved in stimulating the degradation of soluble substrates and aggregates by the proteasome. Further, we found that HUWE1 selectively recognizes substrates bearing high-density ubiquitin chains and strongly increases their ubiquitylation with very high processivity. I call this enzymatic activity "Ubiquitin-Directed ubiquitin Ligase" (UDL), and the overall process "Ubiquitin Signal Amplification" (UbSA). The UDL activity acts on both soluble and aggregated substrates, and dramatically increases their clearance via the ubiquitin-dependent segregase p97/VCP. Our current results suggest that HUWE1’s UDL activity regulates a number of biological activities, including cell division, apoptosis and stress response.
III. “Commander” of the Proteasome
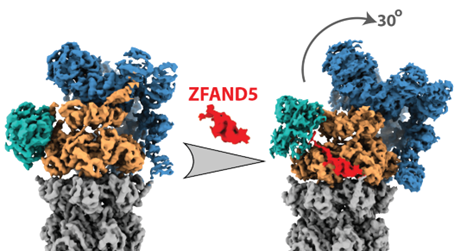
Protein ubiquitylation has long been assumed to be the only regulated step in the ubiquitin-proteasome pathway and the sole determinant of a substrate’s rate of degradation. It is now well established that the capacity of the proteasome system is also tightly regulated and increases in a variety of physiological conditions, such as during heat shock and muscle wasting. However, the mechanism of proteasome activation is largely unclear. In this study, we investigated how ZFAND5, a proteasome cofactor, facilitates proteasome-substrate interaction and synchronizes substrate binding with proteasome conformational cycle to stimulate degradation.
Our team resolved several high-resolution cryo-EM structures of the proteasome-ZFAND5 complex and discovered that ZFAND5 simultaneously interacts with multiple proteasome subunits and triggers a novel conformation of the 19S particle that widens the entrance of the substrate translocation channel. Using single-molecule fluorescence microscopy, we determined how ZFAND5 alters the substrate-processing kinetics of the proteasome. Overall, we found that ZFAND5 acts as a shuttling factor for the 26S proteasome: it binds ubiquitylated substrates, prolongs their association with proteasomes, and increases the likelihood that bound substrates commit to degradation. The coupling between ZFAND5 binding and proteasome conformational change synchronizes substrate binding with the proteasome's conformational cycle, promoting translocation-competent conformational states and enhancing the efficiency of substrate capture. This in-depth mechanistic study provides compelling evidence supporting the original hypothesis that proteasome activity can be dynamically regulated by its cofactors. The results have important implications for the biological processes regulated by ZFAND5 and point the way to novel therapeutics by inhibiting ZFAND5 function.
Related publication:
Donghoon Lee, Yanan Zhu, Louis Colson, Xiaorong Wang, Siyi Chen, Emre Tkacik, Lan Huang, Qi Ouyang, Alfred L. Goldberg* and Ying Lu* (2023) Molecular mechanism for activation of the 26S proteasome by ZFAND5. Molecular Cell, Aug 17;83(16):2959-2975.e7
IV. Predicting Proteasome’s Structural Dynamics Using Empirical Free Energy Landscape
The structural information we and others have collected provides valuable insights into the proteasome function, but many important questions remain. One of the most puzzling is how the six proteasomal ATPase subunits coordinate to capture the energy of ATP hydrolysis and drive substrate degradation. This gap exists because we have little information about the structural dynamics of the proteasome. Resolving the dynamics of protein complexes has been a long-standing challenge in structural biology.
In this study, we developed an innovative method to investigate the complex conformational transitions of the proteasome ATPases. Our “empirical free-energy landscape” method integrates structural information, single-molecule kinetics measurements and a priori physical knowledge, using a small set of parameters to define the energy landscape of the proteasomal ATPase complex. The conformational dynamics of the proteasome can then be computed directly as transitions on its energy landscape, which predicts behaviors such as the steady-state conformational distribution, substrate degradation rates in the presence of different nucleotide combinations, and the growth phenotypes of cells with individual ATPase mutations.
Our empirical free-energy landscape model predicts proteasomal behaviors under various conditions. We found that most of these predictions, including some counter-intuitive ones, are quantitatively or semi-quantitatively consistent with results from previous studies and with new data we generated. Overall, this study demonstrated the feasibility of simulating the actions of a large protein complex using its free energy landscape, a task that would require over 1000 years using conventional molecular dynamics simulation methods. Our method provides a coherent interpretation for the proteasomal phenotypes observed in many studies and addresses important mechanistic questions, such as the origin of the ATPase cooperativity and how the ATPases convert chemical energy into mechanical work during substrate translocation.
Related publication:
Rui Fang, Jason Hon, Mengying Zhou and Ying Lu* (2022). An empirical energy landscape reveals the mechanism of proteasome in polypeptide translocation. eLife. 10.7554/eLife.71911
V. Cryo-EM Structures of Human 26S Proteasome
The lack of structural information about the 26S proteasome has greatly hindered the study of ubiquitin-mediated protein degradation and impeded structure-based design of drugs targeting the proteasome. In a series of studies conducted by my lab and collaborators, we used an improved method for proteasome purification and cryo-electron microscopy with unsupervised structural classification, and resolved the first high-resolution structures of human 26S proteasome and the free 19S proteasome, with atomic details in certain regions.
To understand how the ATPases on proteasome drive its conformational changes crucial for the protein degradation, I designed this study to look for different conformations of the proteasome. I purified the human 26S proteasome, used ATP analogs to enrich alternative proteasome conformations and prepared EM grids. About 1 million cryo-EM particles were imaged. Together, we reconstructed seven conformations of the 26S proteasome at near-atomic resolutions, and thus provided insights into how the ATPases control proteasome conformation to drive substrate translocation. Notably, we identified the mechanism that opens the gate to the 20S particle—an important regulatory step controlling proteasome activity—is allosterically regulated by the insertion of the C-terminal motif of the ATPase Rpt5 into a binding pocket in the 20S proteasome during the ATPase conformational change.
Related publications:
Shuobing Chen*, Jiayi Wu*, Ying Lu*, Yong-Bei Ma, Byung-hoon Lee, Zhou Yu, Qi Ouyang, Daniel J. Finley, Marc W. Kirschner, and Youdong Mao (2016). Structural Basis for Dynamic Regulation of The Human 26S Proteasome. PNAS Nov. 15; 113(46): 12991-12996. *equal contribution
Ying Lu*, Jiayi Wu*, Shuangwu Sun, Yong-Bei Ma, Qi Ouyang, Daniel Finley, Marc W. Kirschner, Youdong Mao (2017). Conformational Landscape of the p28-bound Human Proteasome Regulatory Particle. Molecular Cell Jun 26; S1097-2765(17). *equal contribution
Yanan Zhu, Wei Li Wang, Daqi Yu, Qi Ouyang, Ying Lu*, Youdong Mao* (2018). Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nature Communication Apr 10;9(1):1360. * Co-corresponding author
Yuanchen Dong, Shuwen Zhang, Zhaolong Wu, Xuemei Li, Weili Wang, Yanan Zhu, Svetla Stoilova-McPhie, Ying Lu*, Daniel Finley, Youdong Mao (2019). Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature. 565(7737):49-55.
VI. Deciphering the Death Code of Protein Molecules
Related publications:
Ying Lu, Byung-hoon Lee, Randall King, Daniel Finley and Marc W. Kirschner (2015). Substrate Degradation by the Proteasome: A Single-Molecule Kinetic Analysis. Science, 348:1250834.
Ying Lu, Weiping Wang, and Marc W. Kirschner (2015). Specificity of the Anaphase-Promoting Complex: A Single-Molecule Study. Science 348:1248737.
Byung-Hoon Lee, Ying Lu, Miguel A. Prado, Yuan Shi, Shuangwu Sun, Suzanne Elsasser, Marc W. Kirschner, Steven P. Gygi, Randall W. King, and Daniel Finley (2016). USP14 Deubiquitinates Proteasome-bound Substrates that are Ubiquitinated at Multiple Sites. Nature 532:398-401.
Nicholas G. Brown, Ryan VanderLinden, Edmond R. Watson, Florian Weissmann, Alban Ordureau, Kuen-Phon Wu, Wei Zhang, Shanshan Yu, Joseph S. Harrison, Renping Q. Coudevylle, Iain F. Davidson, Ying Lu, Prakash Dube, Michael R. Brunner, Christy R. R. Grace, Darcie J. Miller, Marc A. Jarvis, Masaya Yamaguchi, Peter Y. Mercredi, Sachdev S. Sidhu, Brian Kuhlman, Marc W. Kirschner, J. Wade Harper, Jan-Michael Peters, Holger Stark, Brenda A. Schulman (2016). Dual RING E3 Architectures for Substrate Polyubiquitination Visualized for Anaphase Promoting Complex/Cyclosome. Cell 165(6);1440-53.
Jason Hon, and Ying Lu* (2018). Single-molecule methods for measuring ubiquitination and protein stability. Methods in Enzymology 619: 225-247. *corresponding author *review article
Hundreds of ubiquitylation pathways conjugate complex patterns of ubiquitins on numerous protein substrates to alter their properties, such as stability, localization, activity and interaction partners. Due to the complexity and experimental challenges, we still do not understand the rules by which the proteasome recognizes this ubiquitin code and commits the destined substrate into an irreversible degradation process.
In previous studies, we introduced chemical methods for generating substrates with defined ubiquitin configurations, and developed single-molecule kinetics approaches to illuminate the transient reaction steps and reaction intermediates. We also developed high-throughput methods to identify the ubiquitin configurations on physiological substrates. Powered by these novel technologies, we aim to understand how the ubiquitylation patterns on substrates, through interacting with the multitude of ubiquitin receptors on the proteasome, control the key steps in proteasomal degradation to regulate protein stability.
